Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 44 nº 3 - May / June of 2011
Vol. 44 nº 3 - May / June of 2011
|
CASE REPORT
|
|
Mayer-Rokitansky-Kuster-Hauser syndrome: a case report and literature review |
|
|
Autho(rs): Rodrigo Manfroi Gutsche1; Lucia Antunes Chagas2; Rodolfo Leal2; André Lima da Cunha3; Maria Célia Resende Djahjah4 |
|
|
Keywords: Paramesonephric (Müllerian) ducts; Abnormalities. |
|
|
Abstract: INTRODUCTION
Mayer-Rokitansky-Kuster-Hauser syndrome is an uncommon condition, with an incidence of one in 40005000 female births(1,2), and is the second most frequent cause of primary amenorrhea after gonadal dysgenesis(3). Such a disorder is a form of Müllerian agenesis characterized by vaginal atresia, and uterine/tubal abnormalities which may include absence or hypoplasia of the uterus and Fallopian tubes. The patients present karyotype 46,XX and normal secondary sexual characteristics, as the ovaries are present and functional, but menstruation is absent(4). Such syndrome is classified into three types according to the involvement of structures other than the ones related to the reproductive system. The typical syndrome (type I) is represented by abnormalities restricted to the reproductive system. The second one (type II) is an atypical syndrome, with the presence of asymmetric uterine remnants and abnormal uterine tubes. Such syndrome type may be associated with ovarian disease, congenital renal, bone abnormalities and hearing defects. A third one, the so called MURCS type, involves uterovaginal hypoplasia or aplasia, renal, bone and cardiac malformations(13). Renal malformations include: unilateral agenesis, horseshoe kidney, renal hypoplasia, ectopic kidneys and hydronephrosis. Bone malformations occur particularly in the vertebrae, most commonly with vertebral fusion (particularly cervical vertebrae), Klippel-Feil syndrome and scoliosis. Cardiac alterations and digital alterations such as syndactyly and polydactyly are rarer than those previously mentioned(1). The syndrome etiology remains unknown, but the increased number of cases in familial aggregates raises the hypothesis of a genetic cause(1). The present report describes the case of a female 16-year-old patient with primary amenorrhea and normal secondary sexual characteristics submitted to clinical-radiological investigation and surgical treatment. CASE REPORT Female, 16-year-old patient with primary amenorrhea, with no other clinical disorder. At clinical examination, the patient demonstrated a development of secondary sexual characteristics compatible with her chronological age. At gynecological examination a grooved urethra with elevated edges was observed. Speculum examination was not performed. Transabdominal ultrasonography did not demonstrate the presence of uterus and ovaries in their habitual site, but the study was inconclusive because of technical difficulties. At magnetic resonance imaging, the uterus could not be visualized and the ovaries presented volume, signal and location with no abnormality. Also, the vaginal canal could not be visualized by such imaging method (Figure 1). 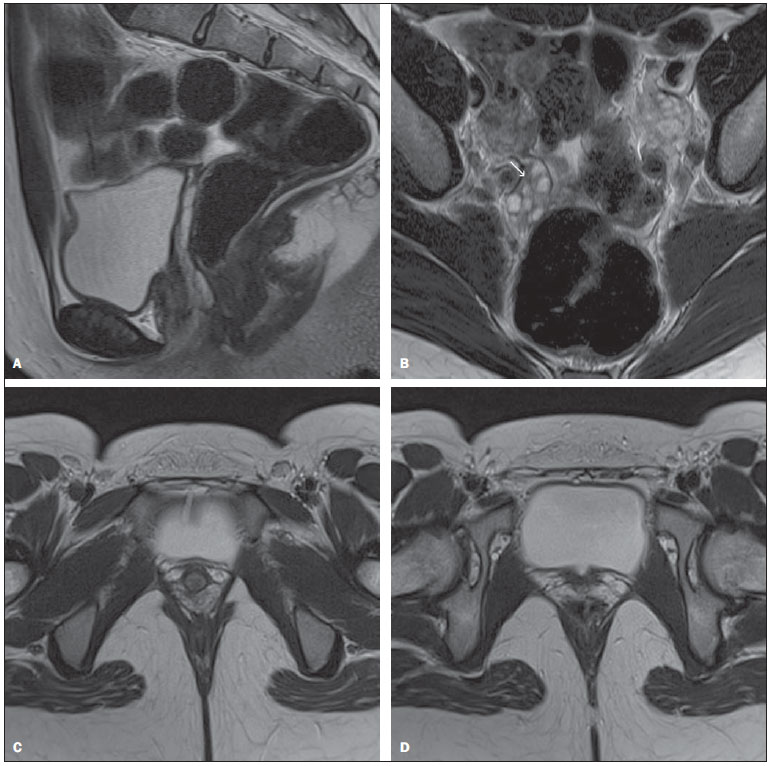 Figure 1. A: Sagittal, MRI T2-weighted image without fat suppression: note that the uterus is not seen in the uterovesical excavation. B: Axial T2-weighted image without fat suppression: note the ovaries with normal volume and shape, in their habitual site, the right ovary presenting follicles (arrow). C: Axial, T2-weighted image: note the absence of the vagina. D: Axial, T2-weighted image: note the absence of the uterus. Genetic evaluation revealed karyotype 46,XX, thus determining the diagnosis of Mayer-Rokitansky-Kuster-Hauser syndrome. The patient was submitted to surgical correction of the vagina (neovaginoplasty). DISCUSSION The typical clinical presentation of this syndrome is primary amenorrhea, in association or not with cyclic colic pain, in an adolescent with secondary sexual characteristics compatible with age, with no sign of virilization. Gynecologic examination may detect either absence of the vaginal canal or vaginal shortening(1,2,58). Imaging studies such as ultrasonography and magnetic resonance imaging, in association or not with laparoscopy, are necessary to allow the determination of the anatomic characteristics of the syndrome. Ultrasonography is the initial method of choice. This method can demonstrate the absence of the uterus between the bladder and the rectum(1,5,9). The vestigial lamina may be confused with the uterus, as it is found in its habitual site. Also, renal anomalies may be observed in cases of type II syndrome(1). Magnetic resonance imaging is the most sensitive and specific imaging method in the evaluation of this syndrome, not only for allowing the acquisition of multiplanar images, but also for allowing the acquisition of sequences with fat saturation. It allows a good definition of anatomical alterations such as uterine agenesis, as well as evaluating ovaries, vagina and associated anomalies(1,4,5,9). Laparoscopy is indicated only in cases where the evaluation by the two previous imaging methods is inconclusive and provided this method allows the definition of a therapeutic strategy. Once the diagnosis of Mayer-Rokitansky-Kuster-Hauser syndrome is established, a clinical investigation should be undertaken to identify possible associated malformations(1,4). The final diagnosis is achieved by the association of the imaging findings with the presence of the karyotype 46,XX. The differential diagnosis should be made with other situations where the patient presents primary amenorrhea and normal secondary sexual characteristics, such as congenital absence of uterus and vagina, isolate vaginal atresia with androgen insensitivity syndrome ans transverse vaginal septum with imperforate hymen(1). Because of the typical anatomic alterations, Mayer-Rokitansky-Kuster-Hauser syndrome generates anxiety and psychological distress with consequences on the patients quality of life, thus requiring a multidisciplinary approach (5,6). The indicated anatomic treatment is the surgical or non‑surgical creation of a neovagina, which may allow these patients to have a normal sex life(16). As the surgical approach is chosen, uterine remnants can be removed to avoid future endometriosis(1). Patients who want to have children should be encouraged to adopt, or the possibility of having biological children by means of assisted reproduction techniques should be suggested, considering that the presence of functional ovaries in these women allow the production of normal ovules(10). Even with the recent developments in the management of this syndrome, its diagnosis causes significant psychological distress, affecting the patients quality of life because of the absence of menstruation and impossibility of pregnancy. The distress caused by the diagnostic may be alleviated by surgical or non-surgical treatments, by the passage of time, by counseling, by familys support and by support groups(10). REFERENCES 1. Morcel K, Camborieux L. Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome. Orphanet J Rare Dis. 2007;2:13. 2. Sultan C, Biason-Lauber A, Philibert P. Mayer-Rokitansky-Kuster-Hauser syndrome: recent clinical and genetic findings. Gynecol Endocrinol. 2009;25:811. 3. Fedele L, Bianchi S, Tozzi L, et al. A new laparoscopic procedure for creation of a neovagina in Mayer-Rokitansky-Kuster-Hauser syndrome. Fertil Steril. 1996;66:854. 4. Sem KK, Kapoor A. Mayer-Rokitansky-Kuster-Hauser syndrome. Ind J Radiol Imag. 2006;16:8057. 5. Junqueira BLP, Allen LM, Spitzer RF, et al. Müllerian duct anomalies and mimics in children and adolescents: correlative intraoperative assessment with clinical imaging. Radiographics. 2009;29:1085103. 6. Muelle GC, Hussain HK, Smith YR. Müllerian duct anomalies: comparison of MRI diagnosis and clinical diagnosis. AJR Am J Roentgenol. 2007;189:1294302. 7. Troiano RN, McCarthy SM. Müllerian duct anomalies: imaging and clinical issues. Radiology. 2004;233:1934. 8. Stainkeler FA, Woodfield CA, Hillstrom MM. Female infertility: a systematic approach to radiologic imaging and diagnosis. Radiographics. 2009;29:135370. 9. Strübbe EH, Willemsen WNP, Lemmens JAM, et al. Mayer-Rokitansky-Kuster-Hauser syndrome: distinction between two forms based on excretory urographic, sonographic, and laparoscopic findings. AJR Am J Roentgenol. 1993;160:3314. 10. Bean EJ, Mazur T, Robinson AD. Mayer-Rokitansky-Kuster-Hauser: sexuality, psychological effects, and quality of life. J Pediatr Adolesc Gynecol. 2009;22:33946. 1. MD, Resident of Radiology at Hospital Universitário Clementino Fraga Filho Universidade Federal do Rio de Janeiro (HUCFF-UFRJ), Rio de Janeiro, RJ, Brazil. 2. Graduate Students of Medicine at Universidade Federal do Rio de Janeiro (UFRJ), Rio de Janeiro, RJ, Brazil. 3. Pediatric Surgeon at Instituto de Puericultura e Pediatria Martagão Gesteira (IPPMG), Rio de Janeiro, RJ, Brazil. 4. MD at the Center of Radiology, Hospital Universitário Clementino Fraga Filho Universidade Federal do Rio de Janeiro (HUCFF-UFRJ), Rio de Janeiro, RJ, Brazil. Mailing Address: Dr. Rodrigo Manfroi Gutsche Rua Conde de Bonfim, 850, ap. 803, Bloco 1, Tijuca Rio de Janeiro, RJ, Brazil, 20530-002 E-mail: rgmanfroi@yahoo.com.br Received October 4, 2010. Accepted after revision February 11, 2011. Study developed at Hospital Universitário Clementino Fraga Filho Universidade Federal do Rio de Janeiro (HUCFF-UFRJ), Rio de Janeiro, RJ, Brazil. |
|
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554