Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 38 nº 6 - Nov. / Dez. of 2005
Vol. 38 nº 6 - Nov. / Dez. of 2005
|
RELATO DE CASO
|
|
|
|
|
Autho(rs): Alexandre Calabria da Fonte, Carlos Marcelo Gonçalves, Julian Catalan, Reinaldo Ottero Justino Junior |
|
|
Descritores: Doença de Camurati-Engelmann, Displasia diafisária progressiva |
|
|
Resumo:
INTRODUÇÃO A doença de Camurati-Engelmann, também conhecida como displasia diafisária progressiva (DDP), é uma síndrome genética autossômica dominante rara, com penetrância altamente variável. Em grande parte dos casos há história familiar da doença, porém casos esporádicos já foram observados(1). A principal característica dessa doença é a formação óssea progressiva endosteal e periosteal, que ocorre principalmente na diáfise dos ossos longos, de forma simétrica, e determina o espessamento cortical, com estreitamento do canal medular e alargamento diafisário(1–3). Clinicamente, os sintomas mais freqüentes são fraqueza muscular e dor nos membros inferiores, porém alguns casos são completamente assintomáticos(1–3). Não existe nenhum teste laboratorial específico para essa doença. O diagnóstico é firmado por meio dos achados clínico-radiológicos(1,2) . Neste trabalho é descrito um caso de doença de Camurati-Engelmann com acometimento ósseo grave.
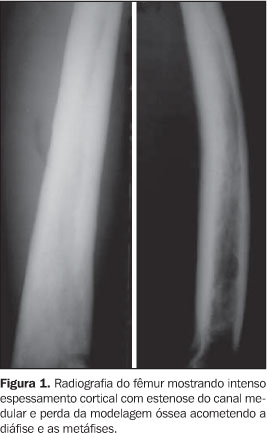
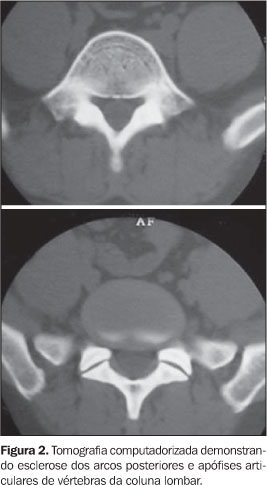
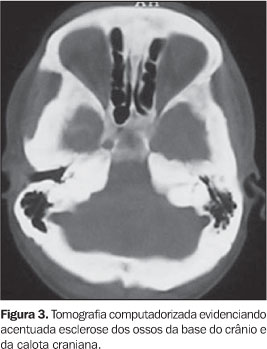
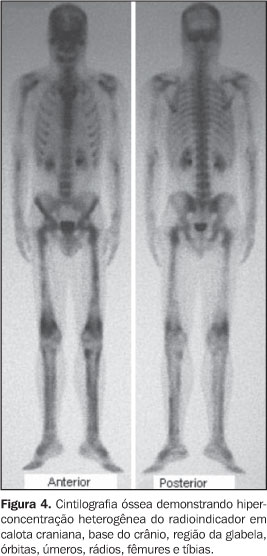
RELATO DO CASO Paciente do sexo masculino, 32 anos de idade, pardo, com queixas de dores generalizadas pelo corpo, mais intensas em membros inferiores, há cerca de nove meses. Não havia história de casos semelhantes na família. O exame físico foi normal. Foram realizadas radiografias simples do esqueleto, que mostraram intenso espessamento cortical, com estenose do canal medular e perda da modelagem óssea, acometendo as diáfises e metáfises dos fêmures, tíbias, fíbulas, úmeros, rádios e ulnas simetricamente (Figura 1). Também foi observada acentuada esclerose da base do crânio, calota craniana, ossos da face e arcos posteriores e processos articulares da coluna lombar. Essas alterações foram confirmadas e melhor observadas na tomografia computadorizada (Figuras 2 e 3). A cintilografia óssea com 99mTc mostrou hiperconcentração heterogênea do radioindicador em calota craniana, base do crânio, região da glabela, órbitas, úmeros, rádios, fêmures e tíbias (Figura 4).
Os achados imagenológicos, associados aos aspectos clínicos e à ausência de anormalidades nos exames laboratoriais, determinaram o diagnóstico de DDP.
DISCUSSÃO A DDP encontra-se no grupo das displasias esclerosantes por defeitos da ossificação intramembranosa, no entanto, em alguns casos ocorre esclerose da base do crânio, que é causada por defeito da ossificação endocondral(1,3). Devido a isso, alguns autores especulam a possibilidade de existirem duas formas de DDP, uma pura, na qual apenas a ossificação intramembranosa é afetada, e outra mista, na qual também há um componente endocondral(1). A sua fisiopatologia ainda não é totalmente compreendida, no entanto, as alterações patológicas parecem ser causadas por uma ativação inadequada da TGFb1, que determina principalmente a redução da atividade osteoclástica, mas também promove aumento da função osteoblástica, favorecendo a deposição óssea(1,2). A apresentação clínica da DDP é bastante inconstante. A idade de aparecimento da doença varia de 3 meses até 50 anos de idade(2). Os sintomas mais freqüentemente encontrados são dores nos membros inferiores, fraqueza muscular e marcha cambaleante. Outros achados menos freqüentes incluem fadiga, atrofia muscular, fraqueza generalizada, exoftalmia, paralisia facial, dificuldade auditiva, perda da visão e puberdade retardada(1–4). Não há nenhuma alteração laboratorial específica. Radiologicamente, observa-se espessamento fusiforme das corticais das diáfises dos ossos tubulares, envolvendo, em ordem decrescente de freqüência, tíbia, fêmur, fíbula, úmero, ulna e rádio(1–4). O acometimento deve ser sempre simétrico e a evolução para a estenose do canal medular é uma constante(1). Outros ossos podem ser acometidos ocasionalmente, como ossos da face, mandíbula, vértebras e a caixa torácica(1,3,4), estes dois últimos somente em casos de doença extremamente grave(3). Apesar de ser doença primariamente diafisária, a progressão metafisária já foi descrita(1–3) e é observada no caso relatado. A tomografia computadorizada do sistema musculoesquelético nos casos de DDP confirma com mais detalhes os achados radiográficos e demonstra a integridade da musculatura adjacente(1). Na cintilografia óssea com 99mTc-MDP é vista captação anormal heterogênea nos ossos afetados, mesmo antes das alterações radiográficas. No entanto, a cintilografia poderá ser normal em alguns casos(1). O diagnóstico diferencial deve ser realizado principalmente com as outras displasias causadas por defeitos na ossificação intramembranosa. As mais importantes são a displasia diafisária múltipla hereditária (doença de Ribbing) e o grupo das hiperostoses endosteais (doença de Van Buchen, doença de Worth, doença de Nakamura e doença de Truswell-Hansen)(1,3). Outras enfermidades que devem ser lembradas são a melorreostose, a displasia craniodiafisária, a doença de Paget, a osteopetrose, a osteoartropatia hipertrófica e a hipervitaminose A(4,5). Os aspectos observados nas radiografias do esqueleto, associados aos achados cintilográficos, são as bases principais para a confirmação diagnóstica da DDP(1,2). Portanto, apesar de ser uma síndrome rara, a DDP é de importância para o radiologista, pois este desempenhará papel significativo no diagnóstico dessa doença.
REFERÊNCIAS 1. Brat HG, Hamoir X, Matthijs P, Lambin P, Van Campenhoudt M. Camurati-Engelmann disease: a late and sporadic case with metaphyseal involvement. Eur Radiol 1999;9:159–162. [ ] 2. Vanhoenacker FM, Janssens K, Van Hul W, Gershoni-Baruch R, Brik R, De Schepper AM. Camurati-Engelmann disease. Review of radioclinical features. Acta Radiol 2003;44:430–434. [ ] 3. Vanhoenacker FM, De Beuckeleer LH, Van Hul W, et al. Sclerosing bone dysplasias: genetic and radioclinical features. Eur Radiol 2000;10:1423–1433. [ ] 4. Lang WS, Viterbo BG, Kiy Y, Gérios JC, Curcelli EC. Displasia diafisária progressiva: aspectos radiológicos e cintilográficos – relato de caso. Rev Imagem 1991;13:17–19. [ ] 5. Greenspan A. Escoliose e anomalias com acometimento geral do esqueleto. In: Greenspan A, editor. Radiologia ortopédica. 3ª ed. Rio de Janeiro, RJ: Guanabara Koogan, 2000;920–924. [ ]
Recebido para publicação em 28/1/2005. Aceito, após revisão, em 18/3/2005.
* Trabalho realizado no Centro de Tratamento e Pesquisa Hospital do Câncer A.C. Camargo, São Paulo, SP. |
|

Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554