Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 51 nº 5 - Set. / Out. of 2018
Vol. 51 nº 5 - Set. / Out. of 2018
|
ENSAIOS ICONOGRÁFICOS
|
|
|
|
|
Autho(rs): Daniel Simões Oliveira; José de Arimatéia Araújo Filho; Antonio Fernando Lins Paiva; Eduardo Seigo Ikari; Rodrigo Caruso Chate; César Higa Nomura |
|
|
Descritores: Pneumonias intersticiais idiopáticas; American Thoracic Society; European Respiratory Society. |
|
|
Resumo: INTRODUÇÃO
As pneumonias intersticiais idiopáticas (PIIs) fazem parte de um amplo e heterogêneo grupo de doenças pulmonares intersticiais que engloba mais de 200 doenças agudas ou crônicas, com graus variados de inflamação e/ou fibrose. Diante da vastidão de aspectos clínicos, radiológicos e patológicos, bem como da constante necessidade de análise evolutiva das PIIs, é imprescindível que o diagnóstico e seguimento dessas doenças sejam firmados em um cenário multidisciplinar no qual o radiologista assume papel fundamental. A tomografia computadorizada de alta resolução do tórax tem sido motivo de recentes publicações na literatura radiológica brasileira(1-7). Não obstante esta abordagem integrada, em alguns casos a classificação final e o diagnóstico definitivo podem não ser alcançados. Tais casos não preenchem critérios para classificação em uma das outras pneumopatias intersticiais clássicas, sendo por isso ditas inclassificáveis e permanecendo como um desafio para toda a equipe envolvida. A última atualização (2013) da classificação das PIIs pela American Thoracic Society/European Respiratory Society(8,9) propõe algumas mudanças importantes em relação à classificação original de 2002. Dentre as novidades, destacam-se a subdivisão das PIIs em quatro grupos principais (crônicas fibrosantes, relacionadas ao tabagismo, agudas/subagudas e raras) e o acréscimo de uma nova doença: a fibroelastose pleuroparenquimatosa idiopática. Neste estudo realizamos uma breve descrição clinicopatológica de cada um desses quatro grupos de PIIs, em paralelo à apresentação de suas principais características radiológicas (Tabela 1), incluindo achados tomográficos, padrões de distribuição e os principais diagnósticos diferenciais. As imagens apresentadas foram selecionadas do arquivo didático de nossa instituição, de casos com padrão tomográfico típico de cada PII, com confirmação anatomopatológica. 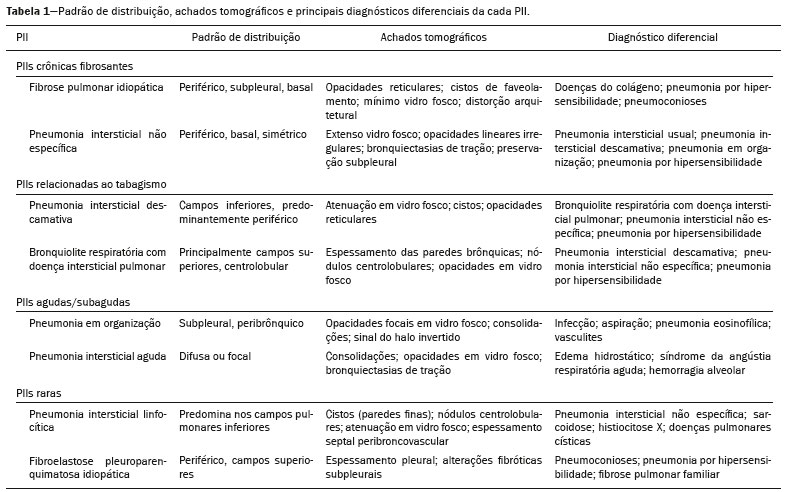 PIIs CRÔNICAS FIBROSANTES Fibrose pulmonar idiopática (Figura 1)  Figura 1. Fibrose pulmonar idiopática. Paciente do sexo masculino, 66 anos, tabagista. Imagem axial de tomografia de alta resolução do tórax (A) e reformatação coronal (B). Em A, opacidades reticulares finas (seta cheia), com bronquiectasias e bronquioloectasias de tração e cistos de faveolamento (seta vazada). Em B, padrão de distribuição predominantemente periférico e basal. Entidade mais comum entre todas as PIIs(10), representa uma pneumopatia intersticial sem causa definida, caracterizada histologicamente pelo padrão de pneumonia intersticial usual(11,12), com focos fibroblásticos dispersos e envolvimento heterogêneo do parênquima, alternando áreas de preservação tecidual com áreas de inflamação intersticial e faveolamento. Ocorre principalmente em adultos fumantes ou ex-fumantes, com idade superior a 50 anos, que apresentam, comumente, dispneia progressiva e tosse seca. Em geral, tem mau prognóstico, com sobrevida estimada em menos de cinco anos a partir do diagnóstico. Os pacientes costumam ter melhora da exacerbação aguda quando tratados com ciclosporina associada a corticosteroides, sendo a maioria deles considerada candidatos a transplante pulmonar após o diagnóstico(10). Em um cenário clínico apropriado (achados clínicos e radiológicos típicos), o diagnóstico de fibrose pulmonar idiopática pode ser firmado sem a necessidade de biópsia(10). Pneumonia intersticial não específica (Figura 2) 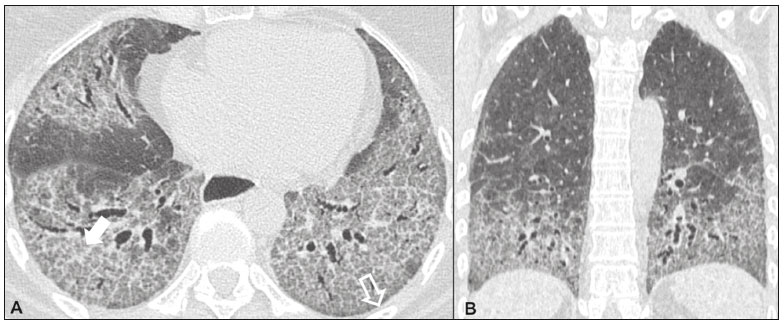 Figura 2. Pneumonia intersticial não específica. Paciente do sexo feminino, 51 anos, com esclerodermia. Imagem axial de tomografia de alta resolução do tórax (A) e reformatação coronal (B). Em A, atenuação em vidro fosco, com opacidades reticulares lineares (seta cheia), bronquiectasias e bronquioloectasias de tração. Nota-se discreta preservação subpleural dos achados (seta vazada). Em B, padrão de distribuição predominantemente basal e simétrico. Caracterizada histologicamente por inflamação homogênea e expansão das paredes alveolares, com ou sem achados de fibrose, pode ser classificada nos subtipos celular (menos comum e com melhor prognóstico), fibrótica (pior prognóstico) ou mista(11). A pneumonia intersticial não específica pode ser idiopática, embora, na maioria das vezes, represente uma manifestação pulmonar associada às doenças do colágeno (sobretudo esclerodermia), à pneumonia por hipersensibilidade, às reações medicamentosas ou ao dano alveolar difuso. Ocorre mais comumente em mulheres entre 40 e 50 anos de idade, com sintomas similares à fibrose pulmonar idiopática, porém, em geral, mais leves. O tratamento é direcionado para a doença de base e pode incluir a associação de corticosteroides sistêmicos e drogas citotóxicas, podendo ser bem sucedido em boa parte dos casos(10). Os achados tomográficos da fibrose pulmonar idiopática e da pneumonia intersticial não específica são muitas vezes similares, cabendo ao radiologista experiente saber as principais diferenças entre elas(13) (Tabela 1). PIIs RELACIONADAS AO TABAGISMO Pneumonia intersticial descamativa (Figura 3) 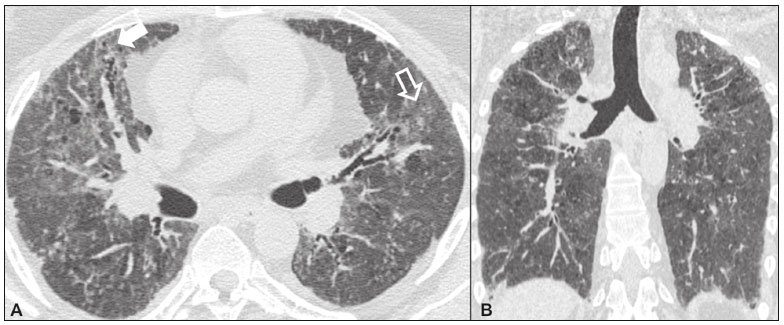 Figura 3. Pneumonia intersticial descamativa. Paciente do sexo masculino, 65 anos, tabagista. Imagem axial de tomografia de alta resolução do tórax (A) e reformatação coronal (B). Em A, opacidades lineares reticulares (seta cheia), com áreas em vidro fosco esparsas bilateralmente (seta vazada). Em B, padrão de distribuição predominantemente periférico. Doença rara fortemente associada ao tabagismo, pode em alguns casos evoluir para insuficiência respiratória e fibrose pulmonar avançada(8). Os achados histopatológicos incluem espessamento homogêneo dos septos alveolares e acúmulo intra-alveolar de macrófagos pigmentados(11). Homens entre 30 e 50 anos de idade compõem a maioria dos pacientes, havendo significativa melhora após cessação do tabagismo e corticoterapia(11). Bronquiolite respiratória com doença intersticial pulmonar (Figura 4) 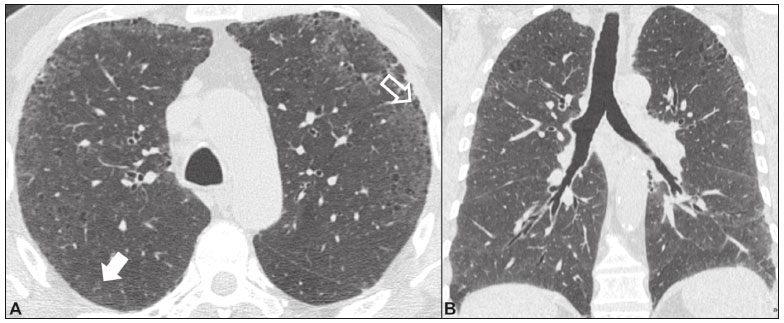 Figura 4. Bronquiolite respiratória com doença intersticial pulmonar. Paciente do sexo masculino, 53 anos, tabagista. Imagem axial de tomografia de alta resolução do tórax (A) e reformatação coronal (B). Em A, opacidades centrolobulares difusas (seta cheia), com espessamento das paredes brônquicas e enfisema parasseptal/centrolobular (seta vazada). Em B, padrão de distribuição predominantemente nos campos superiores. Caracterizada classicamente pela associação de doença intersticial com bronquiolite respiratória, seus achados histológicos típicos incluem o acúmulo de macrófagos pigmentados em bronquíolos respiratórios, ductos alveolares e alvéolos adjacentes, associado a mínima fibrose e infiltrado inflamatório(11). Os pacientes costumam ser homens entre 30 e 50 anos de idade, geralmente oligo ou assintomáticos. O tratamento também consiste em interrupção do tabagismo e corticoterapia(10). PIIs AGUDAS/SUBAGUDAS Pneumonia em organização (Figura 5)  Figura 5. Pneumonia em organização. Paciente do sexo feminino, 62 anos, com dispneia há um ano e piora dos sintomas há dois meses. Imagem axial de tomografia de alta resolução do tórax (A) e reformatação coronal (B). Em A, opacidades em vidro fosco/consolidações esparsas bilateralmente (setas vazadas). Em B, padrão de distribuição subpleural e peribrônquico bilateral. Antigamente conhecida como bronquiolite obliterante com pneumonia em organização, é caracterizada histologicamente por pólipos de tecido de granulação dentro dos ductos alveolares e alvéolos adjacentes, associados a infiltrado inflamatório crônico envolvendo o interstício e os espaços alveolares(8). É um padrão comum de resposta a vários tipos de lesão pulmonar, sendo encontrado mais comumente em associação a doenças do colágeno, reações pulmonares medicamentosas, pneumonia por hipersensibilidade, processos infecciosos e aspiração, entre outros. Quando uma causa não é identificada, denomina-se pneumonia em organização criptogênica(10). Predomina em indivíduos entre 50 e 70 anos de idade, sem predileção por sexo, que apresentam sintomas com duração média de um a três meses, muitas vezes precedida por infecções do trato respiratório(11). O tratamento com corticosteroides costuma ter excelente resposta(9). Pneumonia intersticial aguda (Figura 6)  Figura 6. Pneumonia intersticial aguda. Paciente do sexo feminino, 32 anos, internada por dispneia severa há duas semanas. Imagem axial de tomografia de alta resolução do tórax (A) e reformatação coronal (B). Em A, padrão de atenuação em vidro fosco difuso, notando-se algumas bronquiectasias de tração (seta vazada) e opacidades reticulares (seta cheia). Em B, padrão de distribuição difuso e bilateral simétrico. Doença intersticial aguda idiopática extremamente grave, este tipo de pneumonia caracteriza-se pelo padrão histopatológico de dano alveolar difuso, cuja fase exsudativa é definida por edema intersticial e intra-alveolar, formação de membranas hialinas e infiltração alveolar difusa de células inflamatórias. Apresenta péssimo prognóstico (mortalidade maior que 50%) e normalmente acomete pacientes com idade entre 50 e 60 anos previamente hígidos, causando dispneia severa de instalação abrupta (menos de três semanas), usualmente progredindo para necessidade de ventilação mecânica(10). Os achados clínicos, radiológicos e histológicos são idênticos aos encontrados na síndrome da angústia respiratória aguda(11). O tratamento é basicamente de suporte, com suplementação de oxigênio, sendo eficaz o uso de corticosteroides na fase aguda(10). PIIs RARAS Pneumonia intersticial linfocítica (Figura 7) 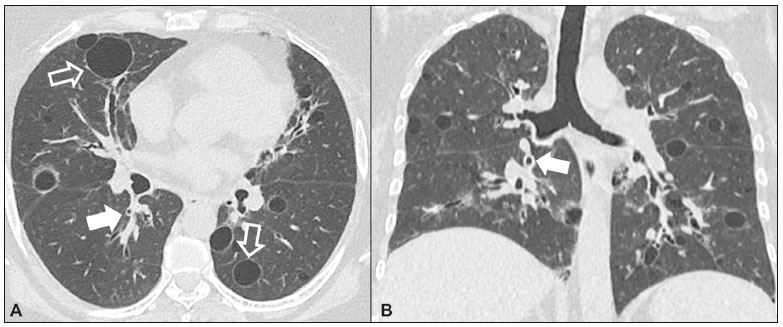 Figura 7. Pneumonia intersticial linfocítica. Paciente do sexo feminino, 62 anos, portadora de síndrome de Sjögren. Imagem axial de tomografia de alta resolução do tórax (A) e reformatação coronal (B). Em A, espessamento difuso das paredes brônquicas (setas cheias), algumas opacidades em vidro fosco e cistos de paredes finas e tamanhos variados, com distribuição difusa e bilateral (setas vazadas). Em B, padrão de distribuição predominantemente nos campos inferiores. Na grande maioria dos pacientes, a pneumonia intersticial linfocítica está associada a alterações sistêmicas autoimunes ou de imunodeficiência, como doenças do colágeno (sobretudo síndrome de Sjögren, tireoidite autoimune e cirrose biliar primária). É considerada extremamente rara na sua forma idiopática. Histologicamente, é caracterizada por infiltração difusa do interstício por linfócitos policlonais, histiócitos e número variável de células plasmáticas, além de folículos linfoides reativos distribuídos pelas regiões peribroncovasculares, com intensa inflamação associada(11). Acomete principalmente mulheres entre 50 e 70 anos de idade, com sintomas inespecíficos de tosse e dispneia crônicas (em geral por mais de três anos). A resposta ao tratamento com corticosteroides é imprevisível(10). Fibroelastose pleuroparenquimatosa idiopática (Figura 8) Figura 8. Fibroelastose pleuroparenquimatosa idiopática. Paciente do sexo masculino, 80 anos, tabagista, com dispneia aos pequenos esforços há um ano. Imagem axial de tomografia de alta resolução do tórax (A) e reformatação coronal (B). Em A, espessamento pleural com alterações fibróticas subpleurais (setas cheias). Em B, padrão de distribuição periférico assimétrico nos campos superiores. Doença rara que afeta a pleura e os pulmões, é caracterizada histologicamente por fibrose subpleural homogênea e fibras elásticas abundantes, com inflamação variável e agregados linfocíticos. Atualmente, não há consenso a respeito dos seus critérios diagnósticos, tampouco sobre o fato de se tratar de uma entidade nova(14). Com pouco mais de 40 casos descritos na literatura até o presente momento, esta entidade tem sido descrita em associação com infecções, transplante de medula óssea, doenças autoimunes e predisposição genética. Seus sintomas são inespecíficos (tosse seca e dispneia) e acredita-se que sua evolução seja lentamente progressiva (10 a 20 anos), com opções terapêuticas limitadas além do transplante pulmonar(15). CONCLUSÃO O correto diagnóstico e classificação das PIIs envolve um amplo debate multidisciplinar, ainda com inúmeras lacunas. Tendo em vista a similaridade dos achados clínicos e anatomopatológicos, seu diagnóstico diferencial complexo e da constante necessidade de acompanhamento evolutivo, a tomografia computadorizada de alta resolução tem papel fundamental na propedêutica dessas doenças. REFERÊNCIAS 1. Torres PPTS, Moreira MAR, Silva DGST, et al. High-resolution computed tomography and histopathological findings in hypersensitivity pneumonitis: a pictorial essay. Radiol Bras. 2016;49:112-6. 2. Mogami R, Goldenberg T, Marca PGC, et al. Pulmonary infection caused by Mycobacterium kansasii: findings on computed tomography of the chest. Radiol Bras. 2016;49:209-13. 3. Queiroz RM, Gomes MP, Valentin MVN. Pulmonary paracoccidioidomycosis showing reversed halo sign with nodular/coarse contour. Radiol Bras. 2016;49:59-60. 4. Koenigkam-Santos M, Cruvinel DL, Menezes MB, et al. Quantitative computed tomography analysis of the airways in patients with cystic fibrosis using automated software: correlation with spirometry in the evaluation of severity. Radiol Bras. 2016;49:351-7. 5. Bastos AL, Corrêa RA, Ferreira GA. Tomography patterns of lung disease in systemic sclerosis. Radiol Bras. 2016;49:316-21. 6. Francisco FAF, Rodrigues RS, Barreto MM, et al. Can chest high-resolution computed tomography findings diagnose pulmonary alveolar microlithiasis? Radiol Bras. 2015;48:205-10. 7. Alves UD, Lopes AJ, Maioli MCP, et al. Changes seen on computed tomography of the chest in mildly symptomatic adult patients with sickle cell disease. Radiol Bras. 2016;49:214-9. 8. Sverzellati N, Lynch DA, Hansell DM, et al. American Thoracic Society-European Respiratory Society classification of the idiopathic interstitial pneumonias: advances in knowledge since 2002. Radiographics. 2015;35:1849-71. 9. Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733-48. 10. Mueller-Mang C, Grosse C, Schmid K, et al. What every radiologist should know about idiopathic interstitial pneumonias. Radiographics. 2007;27:595-615. 11. Silva CIS, Jasinowodolinski D, Terra Filho M, et al. Atlas e diagnóstico diferencial: tomografia computadorizada de alta resolução do tórax - Jorge Kavakama. 1ª ed. Rio de Janeiro, RJ: Revinter; 2008. 12. Lynch DA, Travis WD, Müller NL, et al. Idiopathic interstitial pneumonias: CT features. Radiology. 2005;236:10-21. 13. Silva CIS, Müller NL, Lynch DA, et al. Chronic hypersensitivity pneumonitis: differentiation from idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia by using thin-section CT. Radiology. 2008;246:288-97. 14. Frankel SK, Cool CD, Lynch DA, et al. Idiopathic pleuroparenchymal fibroelastosis: description of a novel clinicopathologic entity. Chest. 2004;126:2007-13. 15. Piciucchi S, Tomassetti S, Casoni G, et al. High resolution CT and histological findings in idiopathic pleuroparenchymal fibroelastosis: features and differential diagnosis. Respir Res. 2011;12:111. Instituto do Coração do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (InCor/HC-FMUSP), São Paulo, SP, Brasil Correspondência: Dr. Daniel Simões Oliveira InCor/HC-FMUSP - Radiologia e Diagnóstico por Imagem Avenida Doutor Enéas de Carvalho Aguiar, 44, Pinheiros São Paulo, SP, Brasil, 05403-900 E-mail: danieloliveira8@live.com Received July 27, 2016 Accepted after revision January 05, 2017 Trabalho realizado no Instituto do Coração do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (InCor/HC-FMUSP), São Paulo, SP, Brasil |
|
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554