Radiologia Brasileira - Publicação Científica Oficial do Colégio Brasileiro de Radiologia
AMB - Associação Médica Brasileira CNA - Comissão Nacional de Acreditação
 Vol. 41 nº 3 - May / June of 2008
Vol. 41 nº 3 - May / June of 2008
|
REVIEW ARTICLE
|
|
Intrathoracic manifestations of collagen vascular diseases on high-resolution chest computed tomography |
|
|
Autho(rs): C. Isabela S. Silva, Nestor L. Müller |
|
|
Keywords: Lung, Collagen vascular disease, Interstitial pneumonia, Pulmonary hypertension, Computed tomography |
|
|
Abstract:
IMD, PhD, Research Associate, University of British Columbia, Vancouver General Hospital, Vancouver, Canada
INTRODUCTION Collagen vascular diseases are a group of diseases with specific autoimmune characteristics that affect several organs including the lungs(1,2). Intrathoracic manifestations are frequent and may be asymptomatic or symptomatic, with varied degrees of severity. The pattern and frequency of the intrathoracic involvement depend on the specific type of collagen vascular disease and may include one or more pulmonary compartments such as alveolus, interstitium, vessels, lymphatic tissue, airways and pleura(1-7). The most frequent pulmonary manifestations are diffuse interstitial pneumonias(1,2,6) and pulmonary hypertension(1,2,8) which as a whole represent the main causes of mortality and morbidity in these patients(1,2). It is important to note that pulmonary abnormalities in patients with collagen vascular disease may be due not only to the underlying disease, but also result from its treatment, and include drug reaction and infection by bacteria or opportunistic organisms, such as Pneumocystis jiroveci, and atypical mycobacteria as a result of immunosuppression(1,2). The collagen vascular diseases that most commonly result in interstitial lung disease are rheumatoid arthritis, progressive systemic sclerosis, systemic lupus erythematosus, dermatopolymyositis, mixed connective tissue disease and Sjögren's syndrome(1,2,6). Chest radiography represents the imaging method most frequently utilized in the initial evaluation of intrathoracic manifestations of collagen vascular diseases. Chest radiography, however, has low sensitivity and specificity. High-resolution computed tomography (HRCT) is the method of choice for assessment of pulmonary abnormalities in collagen vascular diseases, offering the best correlation with histologic findings, disease severity, prognosis, evaluation of disease progression, and differential diagnosis(1). The aim of this manuscript is to review the main intrathoracic manifestations of collagen vascular diseases on HRCT.
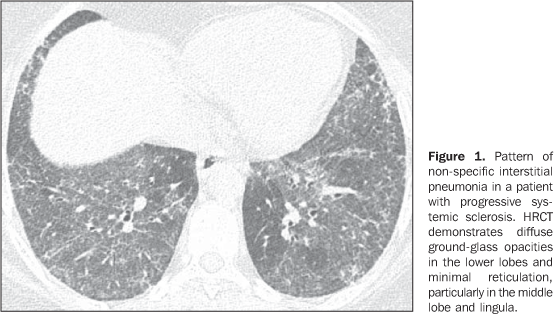
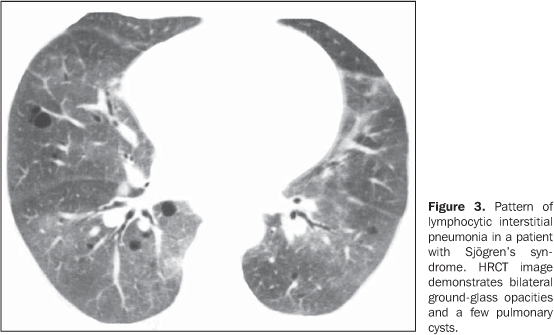
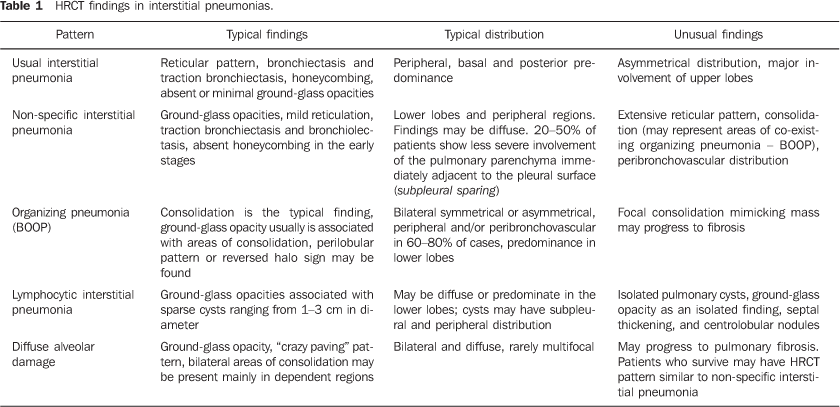
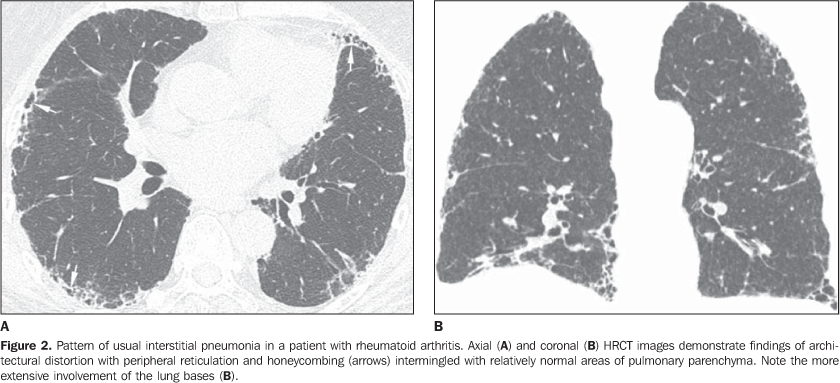
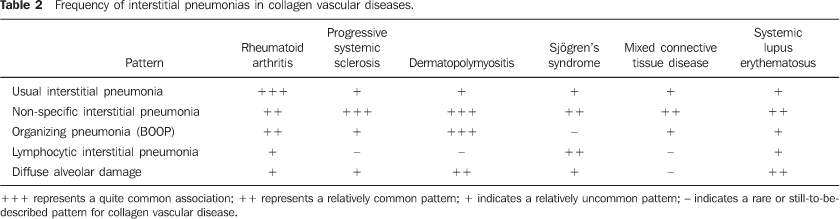
MAIN INTRATHORACIC MANIFESTATIONS OF COLLAGEN VASCULAR DISEASES 1. Diffuse interstitial pneumonias Interstitial pneumonias represent the most common intrathoracic manifestations of collagen vascular disease(1,2). Collagen vascular diseases may be associated with virtually all patterns of diffuse interstitial pneumonia and may progress to pulmonary fibrosis(1-3,6). The histological classification is similar to that of idiopathic interstitial pneumonias including: a) patterns of chronic progression - usual interstitial pneumonia, non-specific interstitial pneumonia and lymphocytic interstitial pneumonia; b) pattern of subacute progression - organizing pneumonia (also known as bronchiolitis obliterans organizing pneumonia —BOOP); c) pattern of acute progression of pulmonary injury - diffuse alveolar damage(3,9,10). High-resolution CT manifestations of interstitial pneumonias in patients with collagen vascular diseases also are similar to the ones described for patients with idiopathic disease (Table 1)(1-3,9,11). Collagen vascular diseases most frequently associated with interstitial pneumonia are: rheumatoid arthritis, progressive systemic sclerosis (scleroderma) and dermatopolymyositis. In general, non-specific interstitial pneumonia is the most common pattern of interstitial pneumonia in patients with collagen vascular diseases (Figure 1)(1,2). In patients with rheumatoid arthritis, however, it seems that the pattern of usual interstitial pneumonia (Figure 2) is the most frequent one(1,7). On the other hand, lymphocytic interstitial pneumonia, although rare, is most common in patients with Sjögren's syndrome (Figure 3)(1,2,12), while the diffuse alveolar damage pattern is more frequent in patients with dermatopolymyositis and systemic lupus erythematosus. Table 2 shows the frequency of each interstitial pneumonia pattern found in the main collagen vascular diseases discussed in the present review.
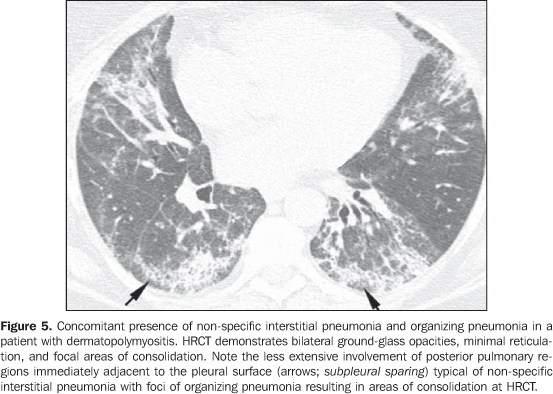
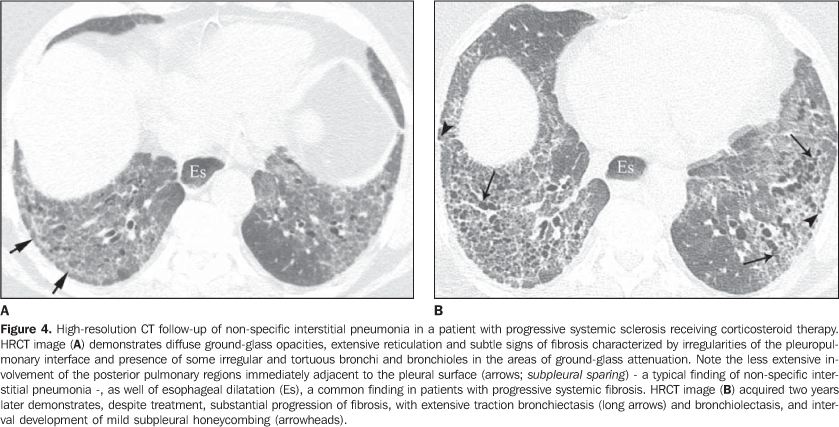
In general, chronic progressive interstitial pneumonias in collagen vascular diseases have a better prognosis than those of idiopathic nature, with a better five year survival rate (Figure 4)(13). However, cases of unfavorable progression in patients with collagen vascular disease and interstitial pulmonary involvement with no response to the therapy are not infrequent. Similarly to the idiopathic form of disease, the non-specific interstitial pneumonia pattern in collagen vascular disease has a better prognosis than the pattern of usual interstitial pneumonia(13). The concomitant presence of more than one pattern of interstitial pneumonia, particularly the association between non-specific interstitial pneumonia and organizing pneumonia (BOOP) is not rare in collagen vascular disease, and is most common in patients with dermatopolymyositis and mixed connective tissue disease (Figure 5). Generally, the pattern of diffuse alveolar damage is associated with poor prognosis, both in patients with idiopathic disease and patients with collagen vascular disease(10).
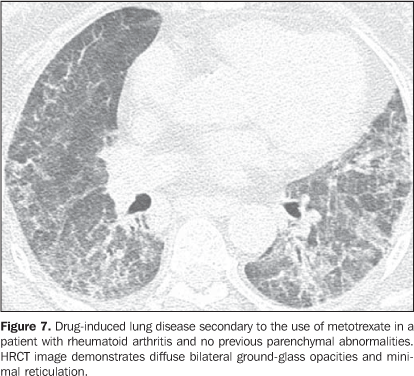
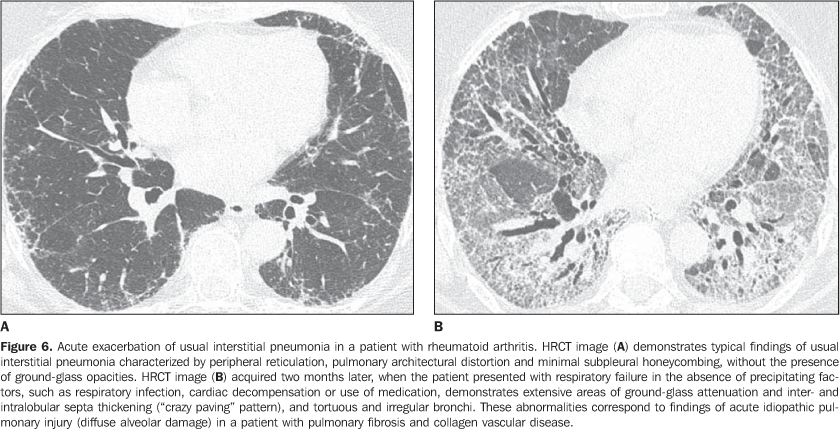
Interstitial pneumonias may precede the clinical onset of the collagen vascular disease for a period of three months up to five years, usually with a pattern of non-specific interstitial pneumonia(14) or acutely with a pattern of diffuse alveolar damage. Patients with collagen vascular disease, especially rheumatoid arthritis, scleroderma and dermatopolymyositis, may progress with acute exacerbation of fibrotic interstitial pneumonia(15). Most frequently, acute pulmonary injury manifests histologically as diffuse alveolar damage superimposed on underlying pulmonary fibrosis (generally usual interstitial pneumonia or non-specific interstitial pneumonia patterns)(15). HRCT findings of acute exacerbation of chronic progressive interstitial pneumonia in patients with collagen vascular disease consist in diffuse areas of ground-glass attenuation, with or without associated smooth inter- and intralobular lines resulting in a crazy paving pattern associated with signs of fibrosis from the pre-existing interstitial pneumonia (distortion of pulmonary architecture, traction bronchiectasis, reticulation and honeycombing) (Figure 6)(15,16). Areas of focal or bilateral dependent consolidation may be present. The main differential diagnosis for ground-glass attenuation at HRCT in patients with collagen vascular disease and fibrotic interstitial pneumonia includes opportunistic infections (particularly by Pneumocystis) and drug-induced lung disease (Figure 7)(1-3). Consolidation may be due to organizing pneumonia or an infectious process (by bacteria, mycobacteria or fungi).
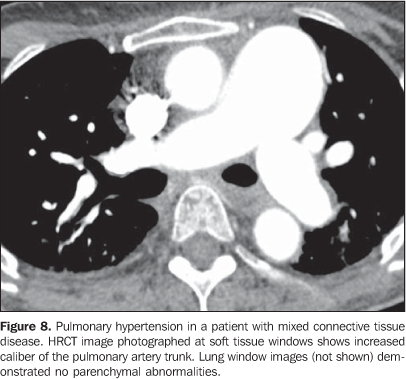
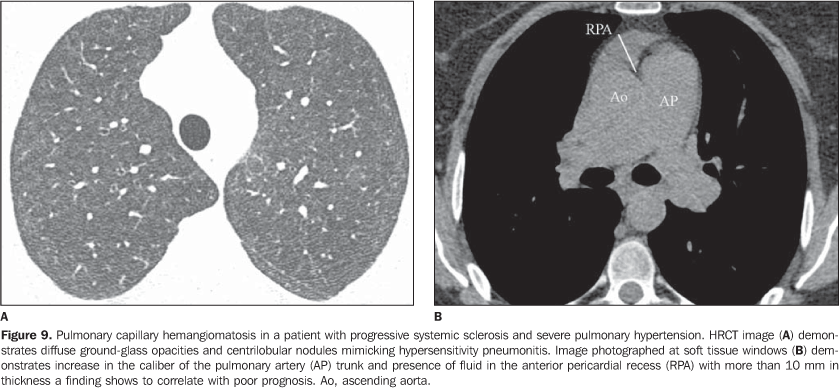
It is important to note that, in smoking patients with collagen vascular disease, smoking-related interstitial pneumonias such as respiratory bronchiolitis with interstitial pulmonary disease and desquamative interstitial pneumonia may also be found(2,9). 2. Pulmonary vascular disease Pulmonary vasculopathy in collagen vascular diseases generally manifests as pulmonary hypertension, diffuse pulmonary hemorrhage (capillaritis) or small and medium vessel vasculitis(2,8). Pulmonary venoocclusive disease and pulmonary capillary hemangiomatosis, although rare, have been described in some patients with collagen vascular disease such as scleroderma and systemic lupus erythematosus(8,17). Pulmonary hypertension in collagen vascular disease may result from a range of different pathophysiological mechanisms including loss of pulmonary capillary network as a result of fibrotic interstitial disease; intrinsic pulmonary arteriopathy with vascular remodelling and pulmonary arterial hypertension; left ventricular diastolic dysfunction with pulmonary venous hypertension; and chronic thromboembolic disease associated with hypercoagulability and antiphospholipides (patients with systemic lupus erythematosus)(8,18). It may occur as an isolated finding (Figure 8) or occurring in association with fibrotic interstitial pneumonia. The incidence of pulmonary hypertension in collagen vascular diseases is variable, depending on the type of collagen vascular disease, on the concomitant presence of interstitial disease and on the diagnostic method utilized(8,18). Pulmonary hypertension is most common in scleroderma (approximately 25% of patients), systemic lupus erythematosus (5-10%) and mixed connective tissue disease(8,18).
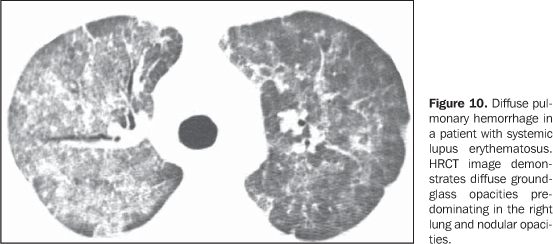
Increase in the diameter of the main pulmonary artery trunk (> 2.9 cm) (Figure 8), main, lobar and segmental pulmonary arteries represent typical findings of pulmonary hypertension at HRCT(3). Artery wall calcification may be identified in patients with severe and longstanding hypertension, as well as thrombi, especially in patients with systemic lupus erythematosus and antiphospholipid antibodies. A mosaic perfusion pattern may be present. Rarely, centrilobular nodules and diffuse or patchy ground-glass opacities are identified at HRCT (Figure 9). These abnormalities may be multifactorial and result from the pulmonary hypertension itself (proliferation of small muscular arteries, chronic passive congestion which may lead to engorgement of the capillary bed, deposition of cholesterol granulomas, focal areas of hemorrhage) or may be associated with the presence of pulmonary capillary hemangiomatosis (Figure 9) (exaggerated multifocal capillary proliferation within the alveolar walls). The concomitant presence of interlobular septal thickening in patients with pulmonary hypertension can be secondary to right cardiac failure or, less frequently, pulmonary venoocclusive disease. The main HRCT manifestations of diffuse pulmonary hemorrhage (capillaritis) in collagen vascular disease consist in diffuse ground-glass opacities (Figure 10), "crazy paving" pattern, patchy consolidation, and centrilobular nodules, or lobular areas of ground-glass opacities(3). The main differential HRCT diagnosis includes: pulmonary infections, especially pneumonia by Pneumocystis, diffuse alveolar damage (Figure 6) and drug-induced lung disease (Figure 7)(1).
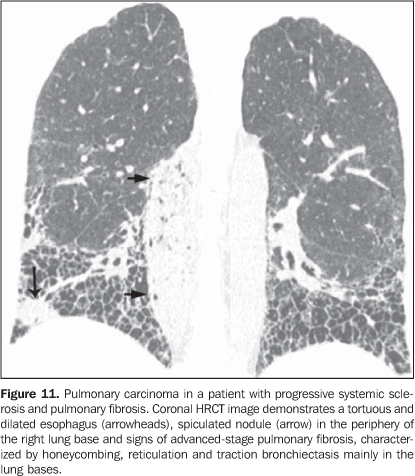
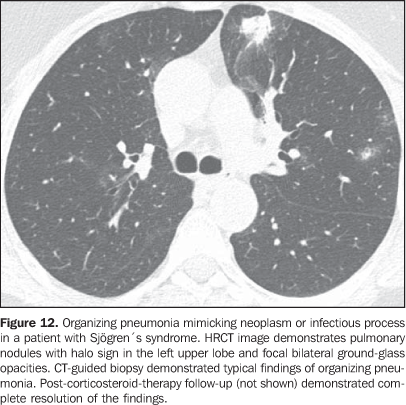
3. Pulmonary nodules or masses The differential diagnosis of pulmonary nodules or masses in patients with collagen vascular disease is broad and includes opportunistic infectious processes (Nocardia, Cryptococcus,Aspergillus, Histoplasma, atypical mycobacteria), neoplasms (carcinoma, lymphoma), metabolic process (amyloidosis), and immunological process (rheumatoid nodules)(19). The main causes of pulmonary nodules and masses found at HRCT of patients with collagen vascular disease are summarized as follows: a) Rheumatoid nodules - Rare, but may be found in patients with rheumatoid arthritis, subcutaneous nodules and positive rheumatoid factor(1,20). Sizes range between 0.5 cm and 7 cm in diameter, and may be single or multiple. These nodules rarely develop in the pleura or pericardium. Rheumatoid nodules may cavitate (up to 50%), causing hemoptysis or pneumothorax, or become infected(1921). Spontaneous resolution may occur. Calcification is rare(19). An unusual association between rheumatoid nodules and pneumoconiosis (particularly in coal-mining workers) in patients with rheumatoid disease is called Caplan's syndrome(19). b) Pulmonary lymphoma - Most frequently occurring in patients with Sjögren's syndrome, it also can be found in patients with rheumatoid arthritis(19,22). At HRCT, pulmonary lymphomas may present as nodules (generally with > 1.0 cm in diameter), consolidation or, rarely, ground-glass opacities(12,22). c) Amyloidosis - The parenchymal nodular presentation of amyloidosis may occur in patients with Sjögren´s syndrome and lymphocytic interstitial pneumonia(22,23). The nodules may be single or multiple, may have foci of calcification and localization in the pulmonary parenchyma or within pulmonary cysts. The diffuse presentation of pulmonary amyloidosis is rare, but may be found in patients with rheumatoid arthritis and Sjögren's syndrome(12, 19,22,24). At HRCT, diffuse pulmonary amyloidosis is characterized by diffuse micronodules and interlobular septal thickening. d) Pulmonary carcinoma - Some studies have shown an increased incidence of pulmonary carcinoma in patients with collagen disease and concomitant fibrotic interstitial vascular disease(1,19); however, this relationship is not well established(25,26). The incidence of neoplasms (including lung cancer) in patients with scleroderma is estimated to range from 3.6% to 10.7%(27). Recently, a study has observed that the incidence of lung cancer in patients with scleroderma is not related to the concomitant presence of pulmonary fibrosis, scleroderma subtype, or to the immunological status of the patient; on the other hand, smoking patients with scleroderma have a seven times higher risk for developing lung cancer than non-smoking patients(28). At HRCT, pulmonary carcinoma may manifest as a spiculated nodule or pulmonary mass (Figure 11). The main differential diagnosis includes rheumatoid nodule (in rheumatoid arthritis), infectious processes (especially fungi), focal amyloidosis and pulmonary lymphoma (particularly in patients with Sjögren's syndrome) and localized organizing pneumonia (BOOP) (Figure 12).
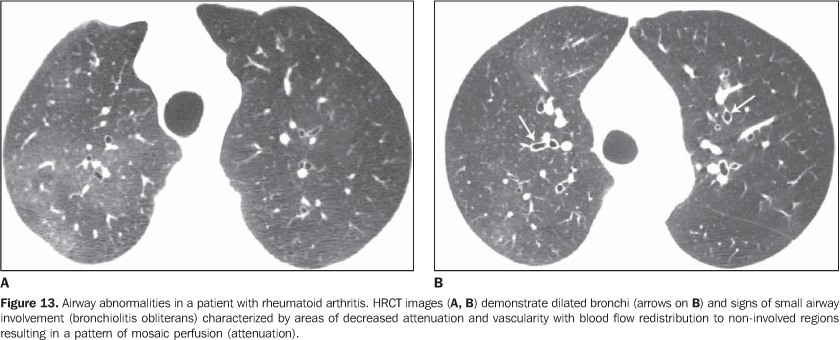
4. Diffuse pulmonary consolidation In patients with systemic lupus erythematosus, diffuse consolidation may result from lupus pneumonitis or diffuse pulmonary hemorrhage (Figure 10)(1,19). Drug-induced lung disease and infection should always be considered in the differential diagnosis. The presence of predominant consolidation in dependent pulmonary regions, particularly in association with centrilobular nodules, may correspond to aspiration pneumonia, a quite common finding in patients with scleroderma and dermatopolymyositis caused by esophageal dysmotility(19,29). 5. Airway a) Large airways - bronchiectasis Bronchiectasis occurs most commonly in rheumatoid arthritis (up to 30%), and also may be found in Sjögren's syndrome and systemic lupus erythematosus(1,3,19). At HRCT, it is more common in the lower lobes, and of cylindrical type (Figure 13)(7). The cause for bronchiectasis in patients with collagen vascular disease is multifactorial; it may be due to repeated infections (particularly in rheumatoid arthritis), involvement of the small airways or airway dryness (Sjögren's syndrome)(1-3,7). b) Small airways-- bronchiolitis - Bronchiolitis obliterans (constrictive bronchiolitis) occurs most commonly in women with rheumatoid arthritis and less frequently in patients with Sjögren's syndrome(1-3). Main HRCT findings include areas of decreased attenuation and vascularity, with or without redistribution of the blood flow towards non-involved regions, resulting in mosaic perfusion pattern (Figure 13)(3,7). Expiratory HRCT demonstrates air trapping. Dilated bronchi (bronchiectasis) (Figure 13) and thickened bronchial walls also can be found(3,7). It is important to note that bronchiolitis obliterans may also result from penicilamine therapy in patients with rheumatoid arthritis(19). Pulmonary hypertension also may result in mosaic perfusion (attenuation) at HRCT; however, at expiratory HRCT, air trapping is not usually found and, differently from bronchiolitis obliterans, an increase in the pulmonary artery caliber is observed. - Follicular bronchiolitis represents bronchiolocentric lymphoid hyperplasia most frequently found in rheumatoid arthritis, Sjögren's syndrome and scleroderma(1-3,30). The HRCT may be normal or show centrilobular and peribronchovascular nodules(31). The main differential diagnosis at HRCT includes infectious process and aspiration (most frequently found in dependent pulmonary regions). - Exudative bronchiolitis (cellular), generally of infectious nature, characterized at HRCT as centrilobular nodules or branching nodular opacities (tree-in-bud pattern) with bilateral and asymmetrical distribution(3). 6. Other intrathoracic manifestations of collagen vascular diseases Collagen vascular diseases may also be associated with other intrathoracic abnormalities summarized as follows: a) Lymphonodomegaly Enlargement o mediastinal lymph nodes is frequently found in patients with collagen vascular disease; it is most commonly associated with the presence of interstitial lung disease, and is found in approximately 70% of patients with scleroderma(19,32). Usually, lymph nodes measure up to 1.5 cm in short axis diameter, and only one or two nodal stations are involved. Enlarged hilar and mediastinal lymph nodes may, however, be found in any patient with collagen vascular disease, including in the absence of interstitial disease, from varied causes such as: sarcoidosis, infection, lymphoma, metastasis, inflammatory response to the presence of reflux or aspiration pneumonia(19,32). b) Abnormalities of the pleura and pericardium Pleural involvement (effusion or thickening) is quite frequent particularly in systemic lupus erythematosus and rheumatoid arthritis(1,19,33). The presence of concomitant pericardial effusion is common. More than half of patients with systemic lupus erythematosus, as the disease progresses, develop pleural effusion, generally bilateral and with a serous or serosanguinolent appearance(33). Pleural effusion, as well as pericardial effusion, may also be a result from the use of procainamide and hydralazine in patients with systemic lupus erythematosus(19). In rheumatoid arthritis, pleural thickening is found in 70% of patients and pleural effusion in approximately 5% of patients(19). In many patients (up to 50%), pericardial abnormalities (pericardial effusion, pericardial adherence and fibrotic or fibrinous pericarditis) are asymptomatic(34). Pericardial effusion, frequently in association with pleural effusion, is one of the most common intrathoracic manifestations of systemic lupus erythematosus. Generally, pericardial effusion has a good prognosis. However, it is important to note that the presence of pericardial effusion associated with pulmonary hypertension (defined by means of echocardiography) in patients with collagen vascular disease, particularly in cases where fluid is detected in the anterior pericardial recess, measuring > 10 mm (Figure 9B), is associated with a poor prognosis(34,35). c) Diaphragmatic dysfunction This is an uncommon condition, but may be found in patients with systemic lupus erythematosus and, occasionally, in patients with dermatopolymyositis(1,3), resulting in dyspnea, restrictive pattern of pulmonary function in the absence of pulmonary abnormalities and diaphragmatic elevation with adjacent basal atelectasis. d) Esophageal dysmotility This is a common condition in patients with scleroderma and dermatopolymyositis(19). Asymptomatic esophageal dilatation may be identified in 40% to 80% of patients with scleroderma (Figure 4)(29). At HRCT, the coronal diameter of the esophageal lumen in patients with scleroderma ranges between 1.2 cm and 4.0 cm (mean 2.3 cm)(29). Esophageal dysmotility may lead to aspiration pneumonia.
CONCLUSION Intrathoracic manifestations of collagen vascular diseases are common and are clinically important. The main pulmonary manifestations include diffuse interstitial pneumonia and pulmonary hypertension which, as a whole, represent the main causes of morbidity and mortality in these patients. The pattern and frequency of parenchymal, vascular, pericardial, pleural and airway involvement, as well as diaphragmatic and esophageal abnormalities depend on the specific type of collagen vascular disease. Awareness of the several patterns of involvement found at HRCT, as well as their clinical/radiological correlation is essential. Intrathoracic manifestations of collagen vascular diseases may be multicompartmental, be due to the underlying disease or be secondary to treatment and thus include drug reaction and opportunistic infection.
REFERENCES 1. Devaraj A, Wells AU, Hansell DM. Computed tomographic imaging in connective tissue diseases. Semin Respir Crit Care Med. 2007;28: 389-97. [ ] 2. Woodhead F, Wells AU, Desai SR. Pulmonary complications of connective tissue diseases. Clin Chest Med. 2008;29:149-64, vii. [ ] 3. Tanaka N, Newell JD, Brown KK, et al. Collagen vascular disease-related lung disease: high-resolution computed tomography findings based on the pathologic classification. J Comput Assist Tomogr. 2004;28:351-60. [ ] 4. Tansey D, Wells AU, Colby TV, et al. Variations in histological patterns of interstitial pneumonia between connective tissue disorders and their relationship to prognosis. Histopathology. 2004; 44:585-96. [ ] 5. Primack SL, Müller NL. Radiologic manifestations of the systemic autoimmune diseases. Clin Chest Med. 1998;19:573-86, vii. [ ] 6. Lamblin C, Bergoin C, Saelens T, et al. Interstitial lung diseases in collagen vascular diseases. Eur Respir J Suppl. 2001;32:69S-80. [ ] 7. Tanaka N, Kim JS, Newell JD, et al. Rheumatoid arthritis-related lung diseases: CT findings. Radiology. 2004;232:81-91. [ ] 8. Hoeper MM. Pulmonary hypertension in collagen vascular disease. Eur Respir J. 2002;19:571-6. [ ] 9. Lynch DA, Travis WD, Muller NL, et al. Idiopathic interstitial pneumonias: CT features. Radiology. 2005;236:10-21. [ ] 10. Won Huh J, Soon Kim D, Keun Lee C, et al. Two distinct clinical types of interstitial lung disease associated with polymyositis-dermatomyositis. Respir Med. 2007;101:1761-9. [ ] 11. Desai SR, Veeraraghavan S, Hansell DM, et al. CT features of lung disease in patients with systemic sclerosis: comparison with idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia. Radiology. 2004;232:560-7. [ ] 12. Parambil JG, Myers JL, Lindell RM, et al. Interstitial lung disease in primary Sjögren syndrome. Chest. 2006;130:1489-95. [ ] 13. Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med. 2007;175:705-11. [ ] 14. Sato T, Fujita J, Yamadori I, et al. Non-specific interstitial pneumonia; as the first clinical presentation of various collagen vascular disorders. Rheumatol Int. 2006;26:551-5. [ ] 15. Silva CI, Müller NL, Fujimoto K, et al. Acute exacerbation of chronic interstitial pneumonia: high-resolution computed tomography and pathologic findings. J Thorac Imaging. 2007;22:221-9. [ ] 16. Horoupian N, English J, Müller NL. Accelerated deterioration of usual interstitial pneumonia with acute development of honeycomb cysts in rheumatoid arthritis. J Thorac Imaging. 2004;19:127-30. [ ] 17. Kishida Y, Kanai Y, Kuramochi S, et al. Pulmonary venoocclusive disease in a patient with systemic lupus erythematosus. J Rheumatol. 1993; 20:2161-2. [ ] 18. Handa T, Nagai S, Miki S, et al. Incidence of pulmonary hypertension and its clinical relevance in patients with interstitial pneumonias: comparison between idiopathic and collagen vascular disease associated interstitial pneumonias. Intern Med. 2007;46:831-7. [ ] 19. Rockall AG, Rickards D, Shaw PJ. Imaging of the pulmonary manifestations of systemic disease. Postgrad Med J. 2001;77:621-38. [ ] 20. Tanoue LT. Pulmonary involvement in collagen vascular disease: a review of the pulmonary manifestations of the Marfan syndrome, ankylosing spondylitis, Sjögrens syndrome, and relapsing polychondritis. J Thorac Imaging. 1992;7:62-77. [ ] 21. Kobayashi T, Satoh K, Ohkawa M, et al. Multiple rheumatoid nodules with rapid thin-walled cavity formation producing pneumothorax. J Thorac Imaging. 2005;20:47-9. [ ] 22. Jeong YJ, Lee KS, Chung MP, et al. Amyloidosis and lymphoproliferative disease in Sjögren syndrome: thin-section computed tomography findings and histopathologic comparisons. J Comput Assist Tomogr. 2004;28:776-81. [ ] 23. Desai SR, Nicholson AG, Stewart S, et al. Benign pulmonary lymphocytic infiltration and amyloidosis: computed tomographic and pathologic features in three cases. J Thorac Imaging. 1997;12: 215-20. [ ] 24. Sumiya M, Ohya N, Shinoura H, et al. Diffuse interstitial pulmonary amyloidosis in rheumatoid arthritis. J Rheumatol. 1996;23:933-6. [ ] 25. Pearson JE, Silman AJ. Risk of cancer in patients with scleroderma. Ann Rheum Dis. 2003;62:697-9. [ ] 26. Hill CL, Nguyen AM, Roder D, et al. Risk of cancer in patients with scleroderma: a population based cohort study. Ann Rheum Dis. 2003;62: 728-31. [ ] 27. Wooten M. Systemic sclerosis and malignancy: a review of the literature. South Med J. 2008;101: 59-62. [ ] 28. Pontifex EK, Hill CL, Roberts-Thomson P. Risk factors for lung cancer in patients with scleroderma: a nested case-control study. Ann Rheum Dis. 2007;66:551-3. [ ] 29. Bhalla M, Silver RM, Shepard JA, et al. Chest CT in patients with scleroderma: prevalence of asymptomatic esophageal dilatation and mediastinal lymphadenopathy. AJR Am J Roentgenol. 1993;161:269-72. [ ] 30. Hayakawa H, Sato A, Imokawa S, et al. Bronchiolar disease in rheumatoid arthritis. Am J Respir Crit Care Med. 1996;154:1531-6. [ ] 31. Howling SJ, Hansell DM, Wells AU, et al. Follicular bronchiolitis: thin-section CT and histologic findings. Radiology. 1999;212:637-42. [ ] 32. Wechsler RJ, Steiner RM, Spirn PW, et al. The relationship of thoracic lymphadenopathy to pulmonary interstitial disease in diffuse and limited systemic sclerosis: CT findings. AJR Am J Roentgenol. 1996;167:101-4. [ ] 33. Kim JS, Lee KS, Koh EM, et al. Thoracic involvement of systemic lupus erythematosus: clinical, pathologic, and radiologic findings. J Comput Assist Tomogr. 2000;24:9-18. [ ] 34. Fischer A, Misumi S, Curran-Everett D, et al. Pericardial abnormalities predict the presence of echocardiographically defined pulmonary arterial hypertension in systemic sclerosis-related interstitial lung disease. Chest. 2007;131:988-92. [ ] 35. Baque-Juston MC, Wells AU, Hansell DM. Pericardial thickening or effusion in patients with pulmonary artery hypertension: a CT study. AJR Am J Roentgenol. 1999;172:361-4. [ ] Received May 5, 2008. Accepted after revision May 27, 2008. * From the Department of Radiology, University of British Columbia, Vancouver General Hospital, Vancouver, Canada. |
|

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Av. Paulista, 37 - 7° andar - Conj. 71 - CEP 01311-902 - São Paulo - SP - Brazil - Phone: (11) 3372-4544 - Fax: (11) 3372-4554